Details about CFEA pipeline

Pipelines
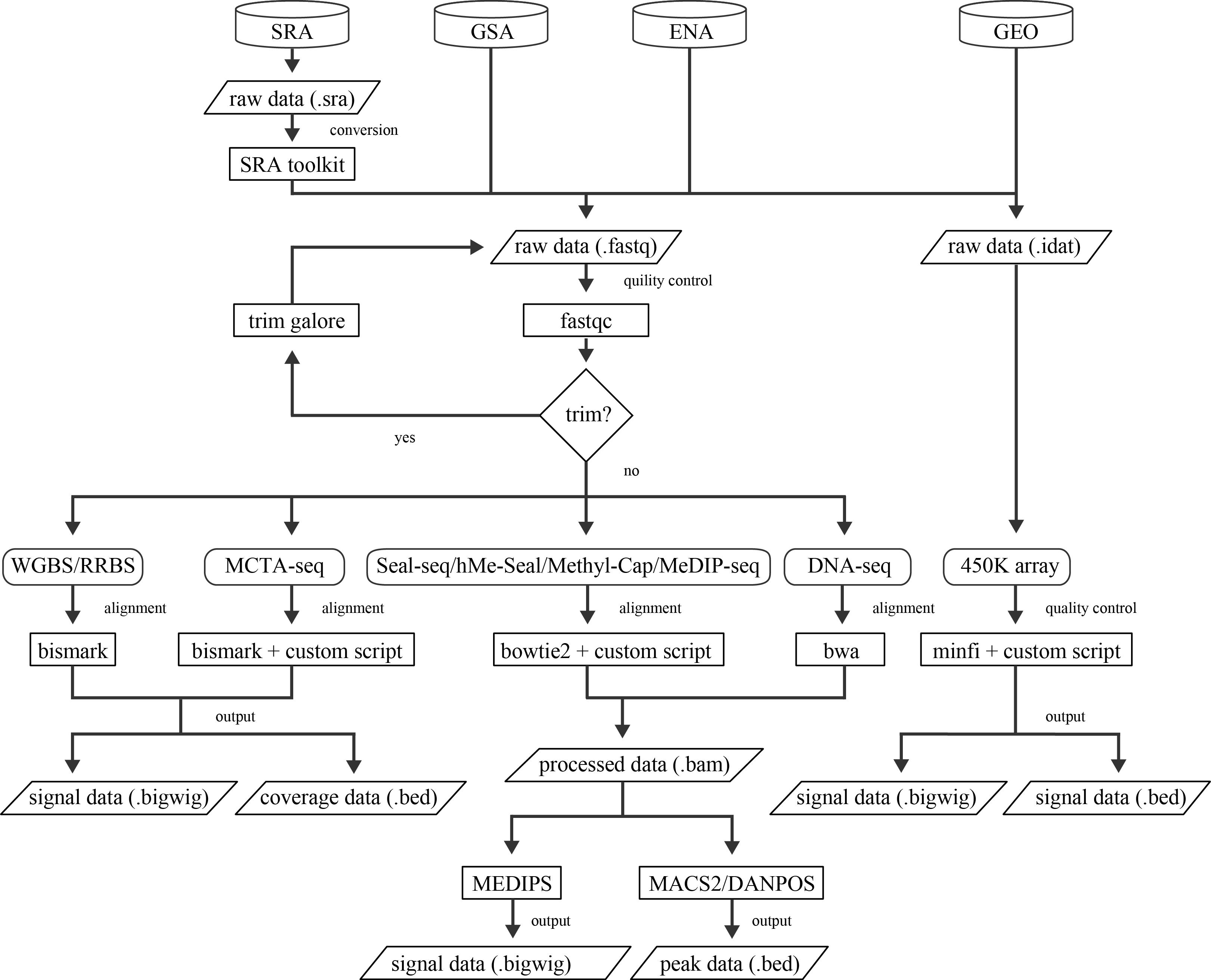
Overview of Pipelines
 Reference:
Reference:
All the detection technologies (9 types) were covered by our pipelines except cMethDNA assay (a low-throughput DNA methylation detection experiment) and we directly downloaded the beta-value-based profiles from GEO web page. Detailed descriptions of the usage are shown below:
Shen S, Singhania R, Gordon C, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes[J]. Nature, 2018, 18(2): 237.
Guo S, Diep D, Plongthongkum N, et al. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA[J]. Nature genetics, 2017, 49(4): 635.
Li W, Zhang X, Lu X, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers[J]. Cell research, 2017, 27(10): 1243.
Wen L, Li J, Guo H, et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients[J]. Cell research, 2015, 25(11): 1250.
Aryee M J, Jaffe A E, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays[J]. Bioinformatics, 2014, 30(10): 1363-1369.
Krueger F, Andrews S R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications[J]. Bioinformatics, 2011, 27(11): 1571-1572.
Software Requirements
We recommend you to install all the softwares into a local environment before starting the data analysis. Conda may be a good option for this purpose.
conda create -n CFEA python=2.7
conda install -n CFEA fastqc bismark bowtie2
macs2 picard multiqc samtools deeptools
Quality controls
We would like to check the quality of the sequencing data before the alignment step. With the script fastqc_fastq.py, we use software Fastqc to evaluate the quality of the raw fastq sequences and software MultiQC to gather the FastQC results into one reporting file.
python raw_fastq_qc.py -i [path to you fastq files] -p 10 -q [qc result dir]
-l [directory to log files] -n [project name in MultiQC]
After the cleaning step above, we will introduce you how to use our pipeline for different data types from various technologies.
WGBS/RRBS (5mC)
Both whole-genome bisulfite sequencing (WGBS) and reduced representation bisulfite sequencing (RRBS) produce bisulfite-converted sequencing data at single-base resolution, so we use the same processing procedure. For the trimming step, we use the tool trim_galore. After trimming, the data was aligned with bismark. The commands are as follows.
trim_galore --rrbs --illumina --phred33 --paired [fastq_file_1]
[fastq_file_2] -o [clean_out_dir] > [log_file] 2>&1
bismark [bismark_index] --path_to_bowtie [bowtie2_path] --bowtie2 -1
[clean_file_1] -2 [clean_file_2] -o [this_sample_mapping_out]
--temp_dir [temp_dir] > [this_sample_bismark_log] 2>&1
The command of the wrapped python script (mapping_WGBS.py) is as follows.
python mapping_bismark.py -i [path fastq] -p [numer of processes] -m
[mapping result dir] -l [log dir] -b [path to put bam files]
-n [project name for multiQC] -t [bowtie2 path] -d [bismark index]
-c [path to put trimmed fastq files]
After the above step, we will get aligned bam files. Next we will use bismark_methylation_extractor to extract methylation coverage from aligned bam files.
bismark_methylation_extractor -s [bam] --bedGraph --counts -o
[this_cov_dir] > [this_log_dir] 2>&1
bismark_methylation_extractor -p [bam] --bedGraph --counts -o
[this_cov_dir] > [this_log_dir] 2>&1
The python script (extract_methylation_coverage.py) to run this command is used below.
python extract_methylation_coverage.py -i [bam dir] -p [number of processed]
-c [coverage out dir] -l [log dir]
The output of bismark_methylation_extractor contains both positive and negative strand CpGs, so we use the below script ro merge these positions.
python merge_cpg_pos.py -i [coverage dir] -p [number of processes]
-f [whole genome CpGs]
Before the above step, you have to get the whole genome CpGs first with script (get_cpg_whole_genome.pl).
perl get_cpg_whole_genome.pl hg19.fa [whole genome CpGs]
Finally, to convert coverage files into wiggle format, simply run.
python coverage_to_wig.py [coverage dir]
Seal-seq/hMe-Seal-seq data (5hmC) & Methyl-Cap/MeDIP-seq data (5mC)
Bowtie2 is used for the aligning purpose and picard is adopted to remove duplicates.
bowtie2 -p 30 --end-to-end -q -x [path_bowtie2_index] -1 [fastq_file_1]
-2 [fastq_file_2] -S [sam_file] > [bowtie2_out_result] 2>&1
samtools view -h [sam_file] | samtools sort -@ 30 -o [sort_bam_file]
picard MarkDuplicates INPUT=[sort_bam_file] OUTPUT=[dedup_bam_file]
METRICS_FILE=[metrics_picard_file] VALIDATION_STRINGENCY=LENIENT
ASSUME_SORTED=true REMOVE_DUPLICATES=true
The wrapped python script runs as follows.
python mapping_5hmc_bowtie2.py -i [path fastq] -p [number of processes] -m
[mapping result dir] -l [log dir] -b [path to put bam files]
-n [project name in MultiQC] -t [bowtie2 path] -d [bowtie2 index]
After deduplicated bams are produced, macs2 is used to call peaks from them.
macs2 callpeak -t [bam] -g hs --nomodel --extsize 200 -n [prefix]
--outdir [this_sample_peak_out] --call-summits > [peak_log] 2>&1
If input data is provided, you may run macs2 like below
macs2 callpeak -t [bam] -c [input bam] -g hs --nomodel --extsize 200 -n [prefix]
--outdir [this_sample_peak_out] --call-summits > [peak_log] 2>&1
Below is our python command.
python callpeak_macs2.py -i [path to bam files] -p [number of processes]
-m [path to put peaks] -l [log dir] -n [multiQC project name]
-o [directory to put peak qc results by MultiQC]
Finally, we can run the below script to make wigs from deduplicated bam files.
bash bam2wig.sh [path to bam files] [path to put wig files]
MCTA-seq data (5mC)
MCTA-seq is a methylated CpG tandems amplification and sequencing method that can detect thousands of hypermethylated CpG islands simultaneously in ccfDNA. The pipeline for MCTA data is a little different from other 5mc data such as RRBS/WGBS. We provide three scripts for the trimming step.
- First we have to trim adaptors and reads with too much N bases as well as too much low quality bases. We have to delete reads with length less than 37 bp after adaptor trimming.
- Second, we have to remove forward 12 bp and backward 6 bp in read1 and forward 6 bp and backward 12 bp in read2.
perl trim_fastq.pl --indir [path to fastq] --outdir [path to put clean fasta]
--sample [sample name]
perl remove_forward6bp.pl [clean fastq 2] [filter fastq 2] 6 20 12
perl remove_forward6bp.pl [clean fastq 1] [filter fastq 1] 12 20 6
After the trimming step, bismark is used to align the reads separately with bowtie1.
bismark --bowtie1 --non_directional --fastq --phred33-quals --temp_dir
[log_dir] --path_to_bowtie [bowtie1_path] --output_dir
[bismark_output_1] [bismark_index] [filter_fq_1] > [bismark_log_1]
2>&1
bismark --bowtie1 --non_directional --fastq --phred33-quals --temp_dir
[log_dir] --path_to_bowtie [bowtie1_path] --output_dir
[bismark_output_2] [bismark_index] [nonCG_fq_2] > [bismark_log_2]
2>&1
The python script covered the above steps runs as follows.
python mapping_MCTA_bismark.py -i [path to fastq files] -p
[number of proceses] -m [path to bismark log files] -l
[path to log files] -b [path to put bam files] -t [bowwtie1 path]
-d [bowtie1 index files] -c [path to trimmmed fastq files]
After the bam files come out, we can do the following steps.
- extract methylation coverage positions from each bam separately with bismark.
- merge CpGs on the positive and negative strand.
- merge two coverage files from tow reads into one coverage file.
The python commands are shown below.
python extract_methylation_coverage.py -i [path to bam files] -p
[number of processes] -c [path to put coverage files] -l [log files]
python merge_cpg_pos.py -i [coverage dir] -p [number of processes]
-f [whole genome CpGs]
python merge_two_coverage.py -i [merged CpG coverage dir] -o
[path to put merged coverage files of two reads]
Finally, to convert coverage files into wiggle format, simply run.
python coverage_to_wig.py [coverage dir]
450K data (5mC)
Parsing 450K data is quite simple, we just follow the minfi tutorial. We also provide a wrapped R script to get the beta values from idat files.
Rscript minfi_beta.R [path to idat files] [path to put beta values]
We also provide you with a script to convert 450K beta values into wiggle format. You have to download the Illumina HumanMethylation450 BeadChip probe annotation file first and parse into the following format.
- CpG probe, such as cg13869341
- chromosome, such as 1, 2, MT...
- genomic positions, such as 15865
The command is shown below.
python beta_450K_to_wig.py [path 450K beta values] [reference file]
DNA-seq (NP)
Nucleosome positioning data is from 2015 Matthew W. Snyder et.al Cell. Fastq files were processed with bwa ALN and sampe command. Aligned reads were stored in BAM format and sorted using the samtools API. Bam files were further processed with Picard to remove duplicates. The commands are as follows.
bwa aln -t 10 -f [bam1] [bwa_index] [fastq1]
bwa aln -t 10 -f [bam2] [bwa_index] [fastq2]
bwa sampe -f [sam] [bwa_index] [bam1] [bam2] [fastq1] [fastq2]
samtools view -h [sam] | samtools sort -@ 10 -o [sort_bam]
picard MarkDuplicates INPUT=[sort_bam] OUTPUT=[dedup_bam_file]
METRICS_FILE=[metrics_picard_file] VALIDATION_STRINGENCY=LENIENT
ASSUME_SORTED=true REMOVE_DUPLICATES=true
The wrapped python script (mapping_np.py) runs as follows.
python mapping_np.py -i [path to fastq files]
-p [number of threads] -m [directory to mapping result]
-l [path to log files] -b [path to put bam files] -d [path to bwa index files]
We use danpos to call peak from deduplicated bam files, and the command to run the python script (call_peak_np.py) is shown below.
python call_peak_np.py -i [directory to bam files]
-p [number of threads] -l [path to log files]
-n [multiQC project name] -o [directory to put peak qc results by MultiQC]
We also provide you with the below script (peak_to_bed_np.py) to convert peak files into bed format file.
python peak_to_bed_np.py -i [peak files] -p [number of processes] -b [directory to put bed files]